对于养细胞的人来说
可谓是谈「污」色变
每次养细胞总让人有种神经错乱
一进细胞房就想让空气都静止下来
打开培养皿的一瞬间
连呼吸都变得如此多余
只要有任何异样
无论是支原体还是杂菌
污染!污染!污染!
犹如细胞实验的魔咒
让一切都无限重复……
那么,如何将失败率降到最低,让我们摆脱玄学的漩涡呢?
今天专门为大家盘点了细胞培养进阶超实用干货,基本的培养和常见问题处理都面面俱到,一篇在手,以后你就能与细胞愉快的玩耍了!
一、细胞污染
#1
如何判断细胞污染了?
现象 1: 细胞培养液变浑浊了,经常见到是米汤样,黄色液体,一般认为细菌污染。

现象 2:细胞培养基内含有能动的小黑点,一般认为是黑胶虫。

现象 3:培养基清亮,但是能看到飘在培养液带有毛毛的白色的菌状物质,有可能就是真菌感染。

解决办法:细胞出现污染就直接丢弃,还得把培养箱用酒精棉球擦干净,并用紫外照射半小时,防止再次污染。同时建议细胞培养时,在培养基中加入青霉素和链霉素(尤其是初学者)
#2
细胞培养过程中,细胞周围包括细胞上有很多小黑点,怎么办?
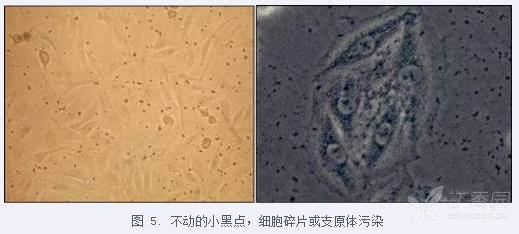
解决办法:细胞出现黑点一般判定为细胞碎片或杂质,可参照以下进行:
如果是悬浮细胞:收集细胞上清慢速离心(500~600 rpm/min,5~6 min)并更换新的培养瓶。
如果是贴壁细胞:将细胞用 PBS 洗 2~3 遍,洗的时候,轻轻拍打培养瓶,让贴壁不牢的碎片和颗粒脱落,再弃去 PBS,消化时先加低浓度的胰酶如 0.05% 胰酶消化 1 min 左右,让细胞间隙中的颗粒和碎片脱落下来,去掉低浓度胰酶,然后正常消化细胞,将收集的细胞悬液慢速离心(500~600 rpm/min,5~6 min)并更换新的培养瓶,并可以尝试适当增加血清浓度进行培养。
如果怀疑黑点是支原体污染,可进行支原体检测,检测阳性请及时将细胞处理后丢弃。比较珍贵的细胞,可以尝试使用支原体清除剂(如泰乐菌素等)去除,并与其他细胞分开培养
如何预防细胞培养中黑点的产生?
解决办法:掌握细胞传代的最佳时机,不要细胞长老了再传代;掌握好消化时间,防止消化过度产生细胞碎片;减少血清等试剂的冻融次数;将培养液的 PH 调到最佳;严格控制水质和器皿的清洁。
#3
细胞培养过程中,发现贴壁细胞表面沾了很多死细胞,传代过程中一直存在怎么破?

解决办法:有一招屡试不爽,那就是用 PBS 洗两遍之后,加胰酶进去,晃一晃,迅速将胰酶倒出,再用 PBS 洗一下,再加胰酶正常消化。死细胞由于粘附能力很弱,第一次加胰酶进去以后会掉下来,第二次加胰酶下来的细胞才是活性比较强的细胞。整完一次之后,再次传代方法同上,一般 2~3 次之后,细胞就完好如初了。
#4
细胞胞质疏松,状态不好,养不起来怎么办?
解决办法1:一般情况下,从血清下手就行,要么换好血清,要么提高血清浓度,血清浓度提高到 15%~20% 培养 48 h,换成正常 10% 的浓度,用高浓度的血清培养时间过长对细胞有毒性。
解决办法2:血清下手之后可以同时采取提高细胞密度的方法,从培养瓶换到六孔板,12 孔板等,细胞密度大,细胞分泌的细胞因子多,肿瘤细胞通过旁分泌途径促进细胞生长。
预防办法:细胞胞质疏松,老化的原因在于细胞传代次数比较多,胰酶消化时间太长,这时候,一定要控制细胞传代次数,注意胰酶消化时间,胰酶消化时间过长,容易导致细胞状态不佳以及胞质疏松。
二、细胞传代
根据细胞生长特性的不同,传代可分为悬浮细胞传代和贴壁细胞传代两种类型。对于悬浮细胞可采用加入等量新鲜培养基后直接吹打分散进行传代,或用离心法后,加入新培养基后再吹打分散进行传代;贴壁生长的细胞则用消化法进行传代,下面我们将介绍传代培养的一些注意事项。
#1
贴壁细胞传代如何使用胰酶?
一般使用 trypsin-EDTA 浓度为 0.25% trypsin-0.53 mM EDTA.2Na 或0.05%trypsin-0.02 mM EDTA.2Na。
消化液浓度过高时,易造成培养基中细胞碎片增多,黑渣子增多,常规细胞传代时建议用 0.05% 的胰酶进行消化,对于难消化的细胞可采用 0.25% 胰酶进行消化,细胞密度过高超过 80% 时,采用分步消化法。
胰酶储存在 –20°C,解冻在 4°C 进行,第一次开瓶后应立即少量分装于无菌试管中,保存于 –20°C,避免反复冷冻解冻造成 trypsin 之活性降低,并可减少污染之机会。
#2
如何控制胰酶消化时间?
胰酶消化过度会导致细胞碎片增多,黑渣子增多,细胞会成片脱落,严重影响细胞活性,并有部分细胞漂浮,随弃去的胰酶流失;消化不足则细胞难以从瓶壁上吹下,反复吹打则会损伤细胞活性。因此,控制胰酶消化时间尤为重要。
不同细胞对消化液的敏感性不同,胰酶消化的时间也会有差异。对于新购买的细胞,建议客户先用低浓度的胰酶仔细去摸索一下消化时间,可每隔 1 分钟镜下观察细胞是否变圆,记录最佳消化时间,下一次操作时参考之前的记录来控制时间即可。
#3
细胞抱团如何处理?
一些悬浮细胞抱团生长是正常现象,大部分悬浮细胞在细胞密度很高的情况下,很可能会出现部分细胞抱团生长的现象,聚团细胞很容易死亡并演化成絮状物,殃及周围的悬浮细胞,因此在培养悬浮细胞时需控制好细胞密度。
如果出现了细胞团,可以通过细胞筛去掉部分较大的细胞团,也可以尝试以下方法:将细胞悬液收集到 15 mL 离心管中,静置 20 min 左右,小心取上层细胞上清培养(该方法只能去除部分较大的细胞团)。
#4
细胞内有空泡,是否是正常现象?
部分细胞本身存在一定的空泡(如 HepG2,Ishikawa 及一些耐药株等),这个是正常现象。如果只有少数细胞有内出现极少空泡,则很可能是细胞状态不佳,可以通过调整血清浓度,控制消化,控制传代比例及时间等方法来调整细胞状态。
如果大部分细胞出现空泡,且单个细胞内空泡数目偏多,则可能细胞代次较高,细胞老化所致,需更换代次较早的细胞。
#5
细胞生长逐渐变慢是什么原因?
l 消化过度
l 传代过密
l 细胞营养不良
l 细胞状态不佳或老化
l 细胞存在污染
#6
细胞不贴壁,有哪些原因?
1)细胞的传代最重要的是胰酶处理时间:消化不够细胞本身就是成团的;若胰酶处理太久,容易造成细胞膜蛋白损伤,使细胞贴壁不紧,显微镜下观察立体感不强,严重的时候甚至会造成细胞死亡。
解决办法:缩短胰蛋白酶消化时间或降低胰蛋白酶浓度。
2)缺少贴壁因子:血清中含有促贴壁因子,而无血清培养基工艺中由于缺少这种促贴壁因子,细胞的贴壁效果会下降很多。
解决办法:更换血清。
3)支原体或细菌的污染。
解决办法:分离培养物,检测支原体。清洁支架或培养箱。如发现支原体污染,丢弃培养物。
4)培养液配置、储存不当,如 pH 值过碱,Gln 量太少等原因。
解决办法:使用无菌醋酸溶液调整 pH 值或充入无菌 CO2。
5)复苏的细胞本身状态不好,已经老化,失去贴附性。
解决办法:启用新的保种细胞。
6)如果细胞比较珍贵或没有备份细胞
解决办法:可尝试将不贴壁细胞收集离心,再用 PBS 将沉淀清洗一遍,离心后的沉淀加入胰酶重新消化接种,同时提高血清浓度到 15%~20%。
三、细胞复苏与冻存

1)取出冻存管,立即放入 37°C 水浴箱中快速解冻(1 分钟左右),水面不可没过盖子,以避免污染。
2)将冻存管移至无菌操作内。打开冻存管,将细胞悬液吸到离心管中,1,000 rpm, 5 min,弃上清。
3)加入适量完全培养基,置于 37°C 恒温箱中培养即可。
4)次日更换一次培养液,以去除任何残留的冷冻保护剂,继续培养。
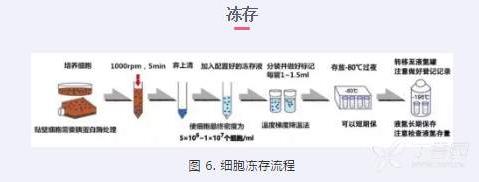
1)用无菌吸管吸弃瓶内旧的培养基;
2)加入少量 PBS 冲洗二遍, 用胰蛋白酶把单层细胞消化下来,依据传代的方法把细胞 悬液收集于离心管中,离心 1,000 rpm,5~8 min;
3)小心弃上清液,加入配置好的冻存培养液,用吸管轻轻吹打使细胞均匀,然后将含有 细胞的冻存液转移到无菌的冻存管中,每个冻存管加液 1~1.5 mL,细胞最终密度为 (5~10)×106 /mL;
4)冻存管封口,做好标记,按照以下程序冻存:
第一种方法:标准的冻存程序为降温速率 -1~-2℃/min;当温度达到 -25℃ 以下时,可增至 -5℃~-10℃/min;到 -150℃ 时,则可迅速浸入液氮中。
第二种方法:冻存管放入 4℃,约 30 min,﹣20℃ 30 min-60 min,见细胞悬液结冻后,放入 -80 ℃ 冰箱, 过夜后转入于液氮罐中。
第三种方法:将冻存管放于程序降温盒中,并置于 -80℃ 冰箱 4 h 以上,转入液氮罐中长期保存。
最后,要着重提醒大家:
除了以上实验操作注意事项,使用合格的细胞产品才是实验成功的第一步。那么,众所周知,真正合格的细胞产品需要具备:细胞种属污染鉴定、细胞支原体阴性,以及最重要的细胞 STR 鉴定。
|
 微信公众号下单更便捷
微信公众号下单更便捷


